SNPlice Usage
Synopsis
Graphical User Interface:
SNPlice
Command-line:
SNPlice [options]
Description
Categorize and count transcript reads (mate-pairs) that span a SNP locus and the transition between an exon and its adjacent intron. Reads are categorized by whether they exhibit the SNP or wild-type allele and whether they are spliced, skipping the intron, or unspliced, reading into the intron. Statistical tests are applied to determine whether the SNP and intron read-through are observed together surprisingly often.
Graphical User Interface
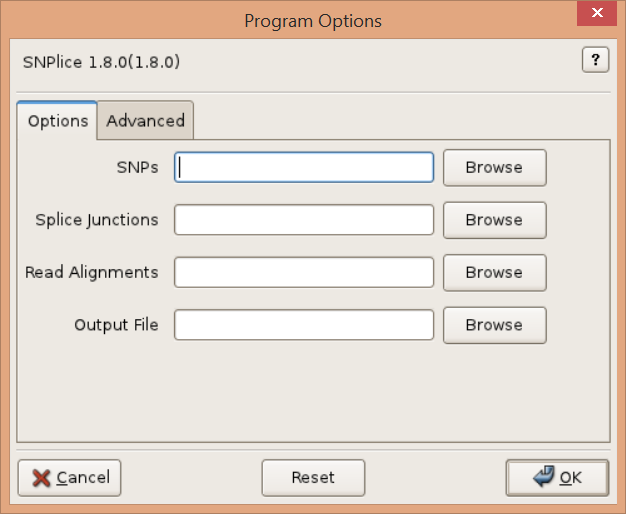
Options
Read Alignments, -r ALIGNMENTS, –readalignments=ALIGNMENTS
Read alignments in (indexed) BAM/SAM format. Required.
SNPs, -s SNPS, –snps=SNPS
Single-nucleotide-polymophisms (SNPs). Tabular and VCF format SNPs are supported. Tabular formats include txt, tsv, csv, xlsx, xls. Text files (txt) must have four spaced separated columns representing the chromosome (CHROM), locus (POS), wild-type allele nucleotide (REF), and SNP nucleotide (ALT). Other tabular formats must provide CHROM, POS, REF, ALT headings. Extra values in tabular or VCF format files are mapped to the output. Required.
Splice Junctions, -j JUNCTIONS, –junctions=JUNCTIONS
Splice junctions in BED format. Junctions are represented as two exon “blocks” separated by an intron, as output by TopHat applied to the reads. Required.
Output File, -o OUTPUT, –output=OUTPUT
Output file. Leave empty for console ouptut. Valid output formats include text (txt extension), tsv, csv, xlsx, and xls. Console output is in text format.
–version
show program’s version number and exit.
-h, –help
Show this help message and exit.
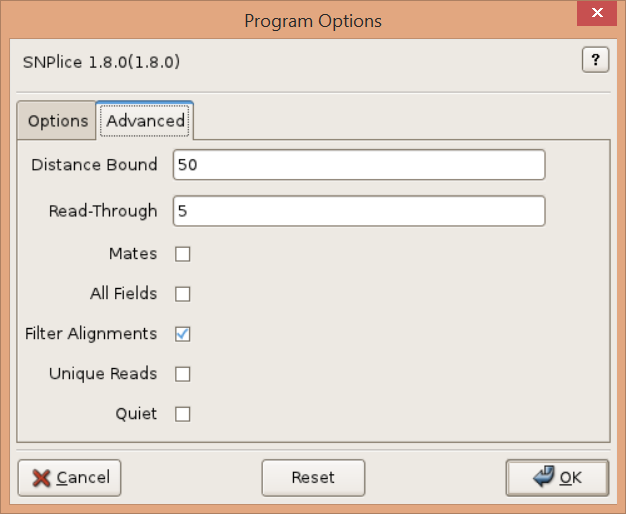
Advanced
Distance Bound, -d DIST, –distance=DIST
Upper bound on the distance between SNP locus and splice junction. Default: 100.
Read-Through, -R READTHROUGH, –readthrough=READTHROUGH
Minimum number of bases required on each side of the intron-exon junction. Default: 5bp.
Mates, -M, –matepairs
Consider read mate-pair alignments to determine splicing status. Default=False.
All Fields, -F, –full
Output extra diagnostic read count fields. Default=False.
Filter Alignments, -f, –alignmentfilter
(Turn off) alignment filtering by length, edits, etc.
Unique Reads, -U, –uniquereads
Consider only reads with distinct sequence.
Quiet, -q, –quiet
Quiet.