RNA2DNAlign Usage
Synopsis
Graphical User Interface:
RNA2DNAlign
RNA2DNAlign.py
Command-line:
RNA2DNAlign [options]
RNA2DNAlign.py [options]
Description
RNA2DNAlign evaluates evidence of allelic imbalance and asymmetry in next-gen sequencing reads of DNA and RNA from normal and tumor samples from the same individual.
Graphical User Interface
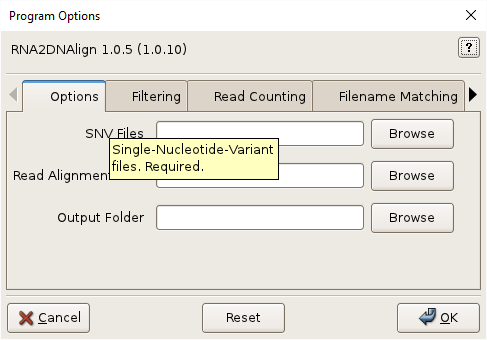
Click the help icon (question mark) at the top right of the GUI and then an input field for help. Multiple files can be selected in the file-chooser using Ctrl-Click or Shift-Click. Fields can be reset to their default values using the Reset button. Click OK to execute RNA2DNAlign.
Additional GUI option tabs are documented below.
Options
SNVs, -s SNVS, –snvs=SNVS
Single-nucleotide-polymophisms (SNVs). Tabular and VCF format SNVs are supported. Multiple files are specified inside quotes, separated by spaces, and by using file globbing. See Input Files for more information. Required.
Read Alignment Files, -r ALIGNMENTS, –readalignments=ALIGNMENTS
Read alignments files in indexed BAM format, with extension
.bam. BAM index with extension.bam.baimust be located in the same directory. Multiple files are specified inside quotes, separated by spaces, and by using file globbing. See Input Files for more information. Required.
Output Folder, -o OUTPUT, –output=OUTPUT
Output directory. Will be created if necessary. Files inside this directory will be overwritten by program output. See Output Files for more information on output files. Required.
–version
Show program’s version number and exit.
-h, –help
Show program help and exit.
Filtering
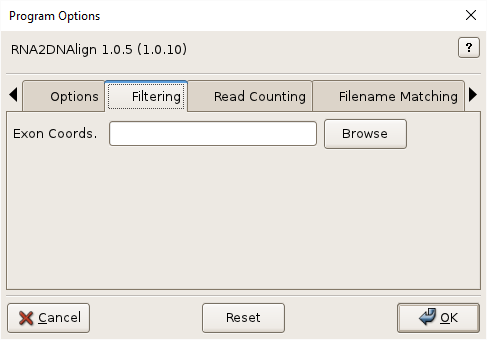
Exon Coords., -e EXONCOORDS, –exoncoords=EXONCOORDS
Exon coordinates to filter out non-exonic SNVs. Use of exon coordinates to filter the SNVs is strongly recommended for transcriptome-to-exome analyses. See Annotation Files for format and download information. Optional.
Read Counting
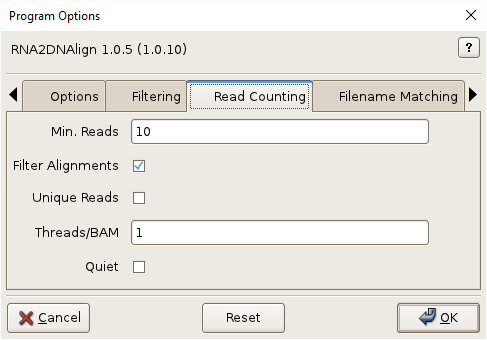
Min. Reads, -m MINREADS, –minreads=MINREADS
Minimum number of good reads at each SNV locus per alignment file. Default=10.
Filter Alignments, -f, –alignmentfilter
(Turn off) alignment filtering by length, edits, etc.
Unique Reads, -U, –uniquereads
Consider only distinct reads.
Threads/BAM, -t TPB, –threadsperbam=TPB
Worker threads per alignment file. Indicate no threading with 0. Default=1.
Quiet, -q, –quiet
Do not show readCounts progress.
Filename Matching
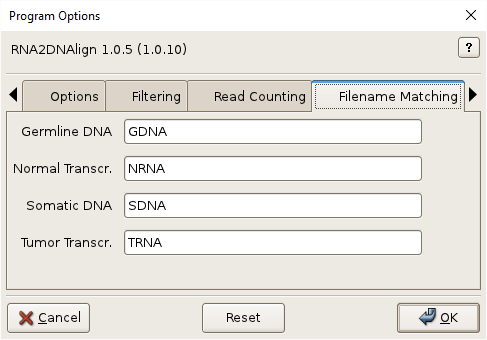
Germline DNA, –normaldnare=NORMALDNARE
Germline/Normal DNA filename regular expression. Default: GDNA.
Normal Transcr., –normaltransre=NORMALTRANSRE
Normal transcriptome filename regular expression. Default: NRNA.
Somatic DNA, –tumordnare=TUMORDNARE
Somatic/Tumor DNA filename regular expression. Default: SDNA.
Tumor Transcr., –tumortransre=TUMORTRANSRE
Tumor transcriptome filename regular expression. Default: TRNA.
SNV Annotation
Darned, -d DARNED, –darned=DARNED
DARNED Annotations. See Annotation Files for format and download information. Optional.
Cosmic, -c COSMIC, –cosmic=COSMIC
COSMIC Annotations. See Annotation Files for format and download information. Optional.
See Also
RNA2DNAlign Home, Input Files, Output Files, Annotation Files, Examples