SCExecute Usage
Synopsis
Graphical User Interface:
scExecute
Command-line:
scExecute -r <bam_file> [options]
Description
SCExecute generates cell-barcode specific BAM files from aligned, aggregate single-cell sequencing data, executing a user-provided command on each barcode-stratified BAM file. Unlike other tools, SCExecute generates cell-barcode specific BAM files in batches to avoid file-system and memory constraints, and manages the execution of the user-provided commands on multiple processors to improve throughput. Cell-barcodes can be extracted from read-names or BAM-file tags populated by a variety of tools, included STARsolo and UMI-tools, and can be restricted to barcodes of interest. Implemented in Python3 using the PySAM package and distributed for Linux, MacOS, and Python environments, SCExecute builds on other NGS tools from the Horvath lab, including SCReadCounts.
Graphical User Interface
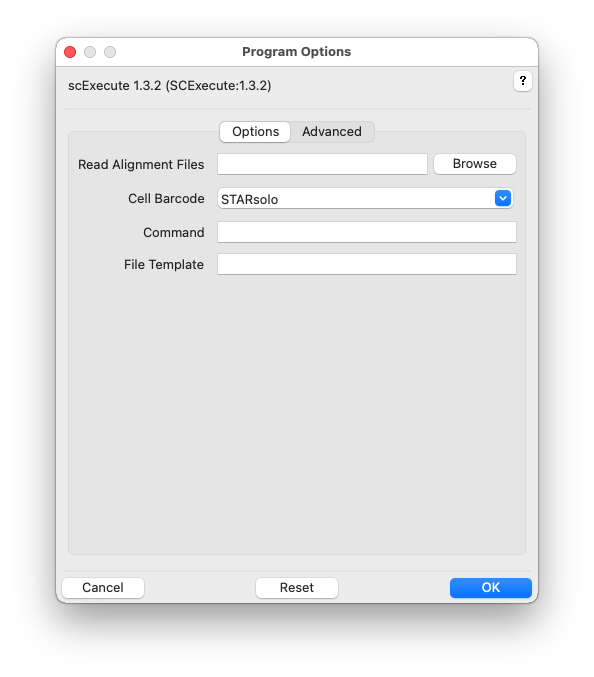
Click the help icon (question mark) at the top right of the GUI and then an input field label for help. Multiple files can be selected in the file-chooser using Ctrl-Click or Shift-Click. Fields can be reset to their default values using the Reset button. Click OK to execute scExecute.
Additional GUI option tabs are documented below.
Options
Read Alignment Files, -r ALIGNMENTS, –readalignments=ALIGNMENTS
Read alignments files in indexed BAM format, with extension
.bam. BAM index with extension.bam.baimust be located in the same directory. Multiple BAM files can be selected from the chooser in the graphical user interface, and on the command-line specified inside quotes, separated by spaces, or by using file globbing. See Input Files for more information. Required.
Cell Barcode, -G READGROUP, –cellbarcode=READGROUP
Cell barcode extraction strategy. Options: Options: CellRanger (Cell barcodes from the CB tag of aligned read - reads without a CB tag or with CB tag not in the accept list (default: file “barcodes.tsv” in the current directory) dropped), STARsolo (Cell barcodes from the CB tag of aligned read - reads without a CB tag or with CB tag not in the accept list (default: file “barcodes.tsv” in the current directory) dropped), UMI-tools (Cell barcodes from read name added by umi_tools). See Cell Barcodes. Default: STARsolo.
Command, -C COMMAND, –command=COMMAND
Command to execute for each cell-barcode specific BAM file. The cell-barcode specific BAM filename replaces {} in the command or is placed at the end of the command if no {} is present. Use {BARCODE} in command as needed, see Command Substitution. At least one of Command/–command/-C or File Template/–filetemplate/-F must be specified.
File Template, -F TEMPLATE, –filetemplate=TEMPLATE
Filename template for each cell-barcode specific BAM file. The cell-barcode specific BAM file template should end in ".bam" and will not be deleted after the command, if specified, is executed. Use {BAMBASE} and {BARCODE} to construct the filename, see Command Substitution. At least one of Command/–command/-C and/or File Template/–filetemplate/-F must be specified.
–version
Show version number and exit.
-h, –help
Show command-help and exit.
Advanced
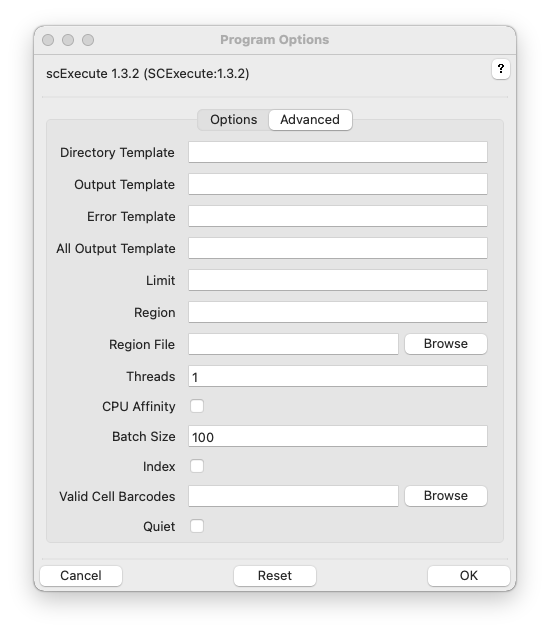
Directory Template, -D TEMPLATE, –directory=TEMPLATE
Working directory for running command on each cell-barcode specific BAM file. Use {BAMBASE} and {BARCODE} to construct the filename, if necessary, see Template Substitution. Default: Current working directory.
Output Template, -o TEMPLATE, –outtemplate=TEMPLATE
Filename template for the standard output of each cell-barcode specific command execution. Use {BAMBASE} and {BARCODE} to construct the filename, see Template Substitution. Default: Standard output of command not captured.
Error Template, -e TEMPLATE, –errtemplate=TEMPLATE
Filename template for the standard error of each cell-barcode specific command execution. Use {BAMBASE} and {BARCODE} to construct the filename, see Template Substitution. Default: Standard error of command not captured.
All Output Template, -O TEMPLATE, –allouttemplate=TEMPLATE
Filename template for the standard output and standard error of each cell-barcode specific command execution. Use {BAMBASE} and {BARCODE} to construct the filename, see Template Substitution. Default: Standard output/error of command not captured.
Limit, -L N, –limit=N
Generate at most
cell-barcode specific BAM files. Default: No limit.
Region, -R REGION, –region=REGION
Restrict reads to those aligning to a single specific region. Default: No restriction.
Region File, –regions
Restrict reads to those aligning to specific region(s). If a GTF-format genome annotation file with extension *.gtf is provided, restrict to genic regions; otherwise, one region per line specified as chrom:start-end in a file with extention *.txt. Default: No restriction.
Threads, -t T, –threads=T
Number of instances of COMMAND to run at once. Default: 1.
CPU Affinity, –cpuaffinity
Constrain each instance of a command to a single CPU. Default: False.
Batch Size, -B B, –batch=B
Number of BAM files to extract with each pass of the input reads. Default: 10.
Index, -i, –index
Index cell-barcode specific BAM file before executing command. Default: False.
Valid Cell Barcodes, -b BARCODES, –barcode_acceptlist BARCODES
File of white-space separated, acceptable cell-barcode values. Overrides filename, if any, specified by Cell Barcode. Use "None" to remove a default accept list.
Quiet, -q, –quiet
Do not show scExecute progress.
See Also
SCExecute Home, Input Files, Cell Barcodes, Command/Template Substitution, Examples