ReadCounts Usage
Synopsis
Graphical User Interface:
readCounts
Command-line:
readCounts -r <bam_file> -s <snv_list_file> [options]
Description
ReadCounts tabulates the number of reads providing evidence for variant and reference nucleotides at specific genomic loci and applies statistical tests to recognize allelic read-counts consistent with homozygous and heterozygous loci.
Graphical User Interface
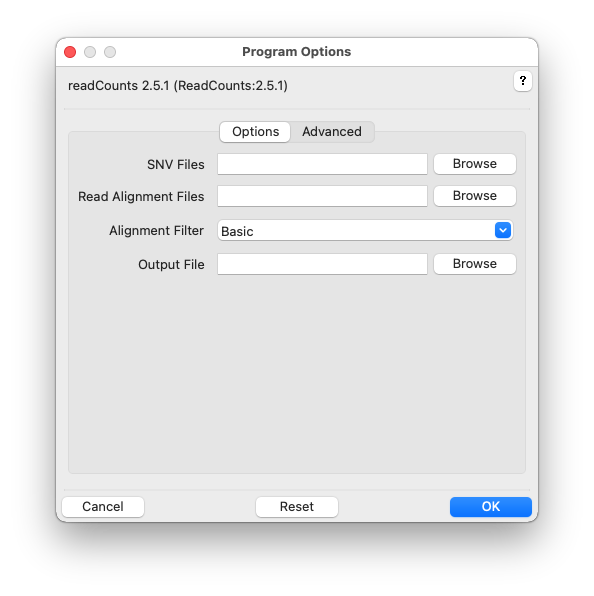
Click the help icon (question mark) at the top right of the GUI and then an input field label for help. Multiple files can be selected in the file-chooser using Ctrl-Click or Shift-Click. Fields can be reset to their default values using the Reset button. Click OK to execute readCounts.
Additional GUI option tabs are documented below.
Options
SNVs, -s SNVS, –snvs=SNVS
Single-nucleotide-variants (SNVs). Tabular and VCF format SNVs are supported. Multiple SNV files can be selected from the chooser in the graphical user interface, and on the command-line specified inside quotes, separated by spaces, or by using file globbing. See Input Files for more information. Required.
Read Alignment Files, -r ALIGNMENTS, –readalignments=ALIGNMENTS
Read alignments files in indexed BAM format, with extension
.bam. BAM index with extension.bam.baimust be located in the same directory. Multiple BAM files can be selected from the chooser in the graphical user interface, and on the command-line specified inside quotes, separated by spaces, or by using file globbing. See Input Files for more information. Required.
Alignment Filter, -f FILTER, –alignmentfilter=FILTER
Alignment filtering strategy. See Read Filtering for more details. Default: Basic.
Output File, -o OUTPUT, –output=OUTPUT
Output file. Will be created if necessary. See Output Files for more information on output files. Optional.
–version
Show version number and exit.
-h, –help
Show command-help and exit.
Advanced
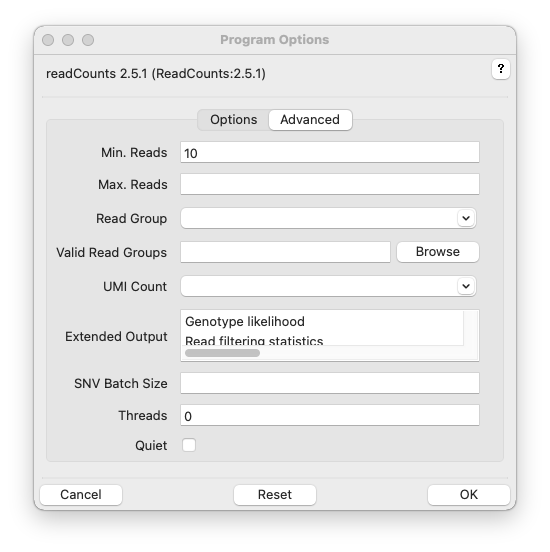
Min. Reads, -m MINREADS, –minreads=MINREADS
Minimum number of good reads at each SNV locus per alignment file. Default=10.
Max. Reads, -m MAXREADS, –maxreads=MAXREADS
Scale read counts at high-coverage loci to ensure at most this many good reads at SNV locus per alignment file. Values greater than 1 indicate absolute read counts, otherwise the value indicates the coverage distribution percentile. Default=No maximum.
Read Group, -G READGROUP, –readgroup=READGROUP
Additional read grouping based on read name/identifier strings or BAM-file RG. See Read Grouping for more details. Default: None, group reads by BAM-file only.
Valid Read Groups, -b BARCODES, –barcode_acceptlist BARCODES
File of white-space separated, acceptable read group values (barcode accept list). Overrides value, if any, specified by Read Group. Use None to remove a default accept list.
UMI Count, -U UMIGROUP, –umicount=UMIGROUP
Count unique identifiers (UMI) based on read name/identifier strings or BAM-file tags. Default: None, count reads not UMIs.
Extended Output, -E, –extended=EXTENDED
Generate extended output, one or more comma-separated values: Genotype likelihood, Read filtering statistics. Default: No extended ouptut.
SNV Batch Size, -B SNVBATCHSIZE, –snvbatchsize=SNVBATCHSIZE
Manage memory footprint by making multiple passes through the BAM file, one for each batch of SNVs. Default=All SNVs (single pass).
Threads, -t THREADS, –threads=THREADS
Worker threads. Default: 0, indicating single-threaded serial execution.
Quiet, -q, –quiet
Do not show readCounts progress.